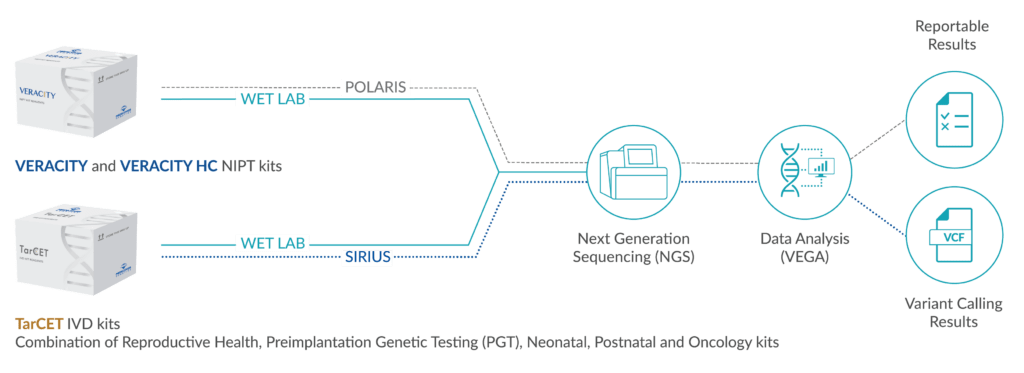
Genetic Testing – Medicover Genetics

WHY MEDICOVER GENETICS?

A leader in genetic testing with
>25 years of experience in counselling
and diagnostics

Provider of turn-key solutions through
CE IVD Kits and Technology Transfer platform for laboratories of any size

Combined with histopathology
and clinical laboratory testing

Strong clinical team providing medical validation and genetic counselling

OUR GENETIC TESTS
We offer genetic testing for different life stage and for predictive and diagnostic testing.
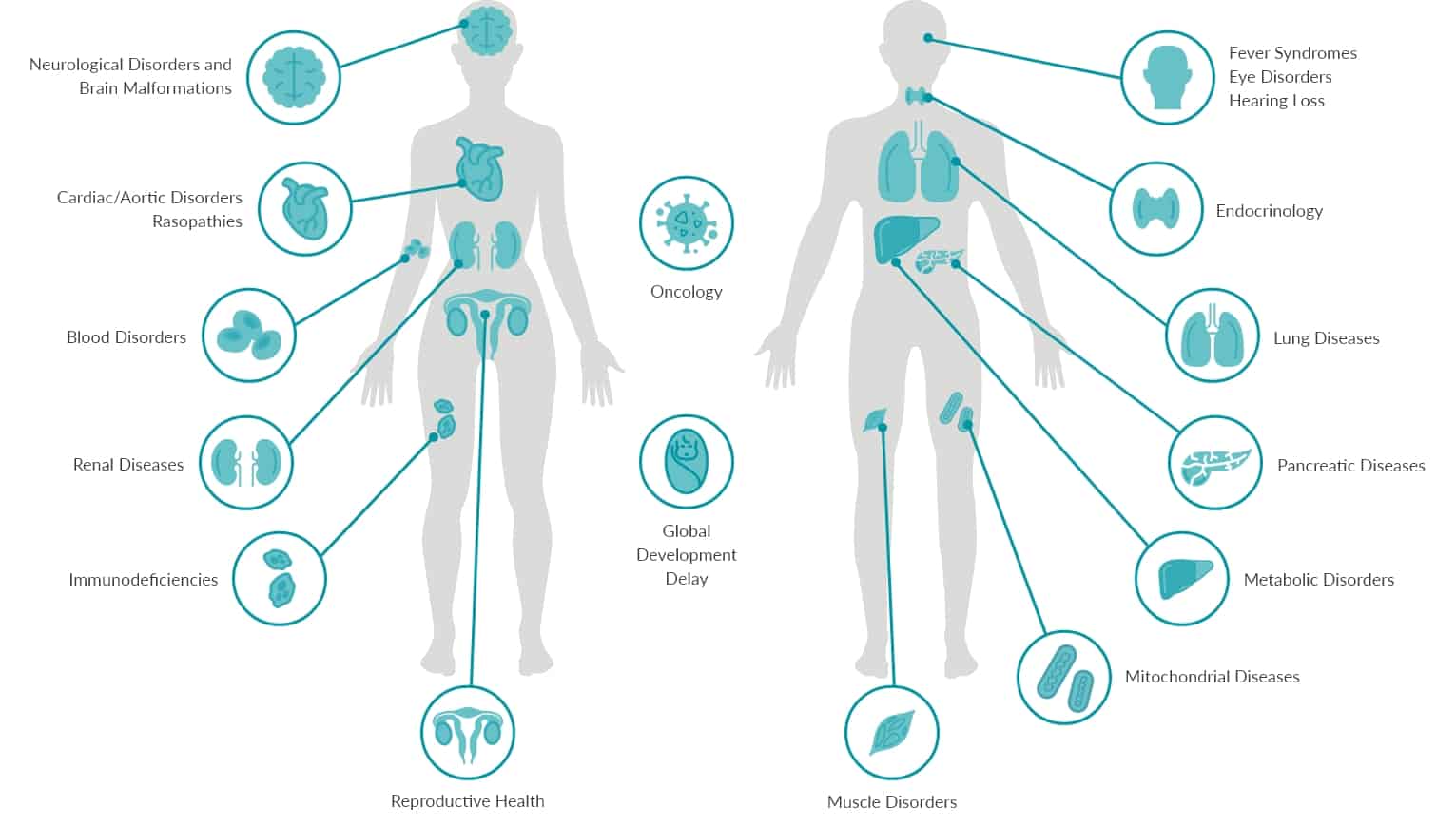

Neurological Disorders and
Brain Malformations

Cardiac/Aortic
Disorders
Rasopathies

Renal Diseases

Blood Disorders

Reproductive

Immunodeficiencies

Oncology

Global
Development
Delay


Fever Syndromes
Eye Disorders
Hearing Loss

Endocrinology

Lung Diseases

Pancreatic
Diseases

Metabolic
Diseases

Mitochondial
Diseases

Muscle Disorders

IS GENETIC TESTING RIGHT FOR YOU?


Genetic tests for cancer
You have a strong family history of cancer or are diagnosed with cancer

Genetic tests for rare diseases
You are a parent of a child with a genetic disorder or with symptoms of unknown origin

Family history of genetic diseases
You have a family history of a genetic disorder

Genetic tests for reproductive health
You are pregnant or interested in conceiving

Our genetic test portfolio
You are a physician and want to find the right test for your patient
LATEST NEWS

Medicover Genetics has successfully acquired MVZ Humangenetik Köln GmbH, expanding its network of genetic counselling centers and laboratories in Ge...

This past year, in an effort to raise awareness about the importance of DNA in modern clinical practice and precision medicine, Medicover Genetics Cy...

Nicosia, Cyprus: Medicover Genetics has successfully renewed its accreditation from the College of American Pathologists (CAP), following a recent on...

Medicover Genetics, Cyprus was honored to host the visit of the Swedish Ambassador to Cyprus, His Excellency Mr. Martin Hagström to our local facili...

This year, MVZ Martinsried celebrates its 25th anniversary, marking a quarter-century of delivering comprehensive and meaningful diagnostic testing t...

Nicosia, Cyprus | October 17, 2023 - Medicover Genetics, a leader in molecular diagnostics with 25 years of experience in genetic medicine, announced...

Last week, Medicover Genetics had the pleasure and honor to attend and present our genetic testing services and laboratory solutions at the 56th conf...

Medicover Genetics and Medicover Diagnostics Finland are proud to announce the partnership with Fimlab Laboratories, to provide genetic testing servi...

Mittwoch 01.03.2023 | 17:00 - 20:00 UhrSehr geehrte Kolleginnen Kollegen,hiermit möchten wir sie herzlich zum 18. Symposium Genetik und Schwangersch...

Following the investment in acquiring NIPD Genetics, Medicover has been recently awarded Invest Cyprus International Investment Awards, a prize honor...
BLOG ARTICLES
Background information on in vitro diagnostic services Laboratory-based testing methods and medical devices play a critical role in diagnosis and ...

Traditional DNA tests may overlook 10% of classic in Familial Adenomatous Polyposis (FAP) cases. By integrating RNA sequencing, researchers unveiled ...

Overview In January 2024, the American Society of Clinical Oncology (ASCO) and the Society of Surgical Oncology (SSO) published new recommendation...

Endometriosis is a chronic gynecological condition that affects 1 in 10 women of reproductive age worldwide [1]. It can manifest with the first menst...

Rare Disease Day is a global awareness day held annually to raise awareness of all rare diseases. It was first celebrated in 2008, on the rarest day ...
Cancer is a group of genetic diseases that can develop almost anywhere in the body. Many people in the world are affected by cancer every year. Follo...

Aiming to evaluate the role of chromosomal aneuploidy in pregnancy loss, a 2023 study 35 years in the making evaluated the genomic landscape of first...

Cancer is a complex genetic disease that affects millions of people in the world. It is one of the leading causes of death worldwide, with about ten ...
Researchers created a detailed map of the placenta during labor. By studying how maternal and fetal cells communicate, they discovered signals in the...
A recent paper published in Nature Medicine (1) aimed to identify genes and genomic biomarkers which can better predict outcomes and personalized the...
HOW TO ORDER A TEST
1
Speak with your physician to find the right test for you or a family member
2
Provide a sample at your nearest blood drawing point
3
Expect the genetic report in 15-25 working days
4
Speak with your physician about the results and next steps
5
Speak with Medicover Genetics counsellors to help explain the results

MEET OUR DIVERSE TEAM IN
12 DIFFERENT COUNTRIES