The main hereditary cardiac arrhythmias are Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT), long QT syndrome (LQTS) and short QT syndrome (SQTS). They share several features, including an overall low prevalence, a difficult diagnosis, and the potential to trigger life-threatening arrhythmias.16 These rare disorders are caused by variants in genes encoding ion channels or proteins involved in their regulation and are often the underlying cause of sudden cardiac death in the young.
Contents
- Main types of hereditary cardiac arrhythmias
- Brugada Syndrome (BrS)
- Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
- Long QT Syndrome (LQTS)
- Short QT Syndrome (SQTS)
- Genetic testing of hereditary arrhythmias
- Recommendations
- Diagnostic Algorithm
- Diagnosis and management
- Case studies
- Brugada syndrome (BrS) case study
- Catecholaminergic polymorphic ventricular tachycardia (CPVT) case study
- Long QT syndrome (LQTS) case study
- Short QT syndrome (SQTS) case study
- References
Main types of hereditary cardiac arrhythmias
Brugada Syndrome (BrS)
Rare variants in the SCN5A gene are found in ~20% of patients with Brugada syndrome.8 Rare variants have also been reported in an additional 21 genes, but SCN5A remains the only undisputed Brugada syndrome-related gene according to systematic assessment by the ClinGen initiative. Other genes have almost exclusively been discovered through candidate gene studies in single individuals or small families. As opposed to LQTS-associated variants, BrS-associated SCN5A variants are loss of function.
The largely sporadic presentation of the disorder and the low disease penetrance in families with rare variants in SCN5A, as well as the observation of phenotype-positive genotype-negative individuals in such families, has suggested that BrS is a disorder with an inheritance more complex than the Mendelian inheritance that was previously considered.15 A genome-wide association study identified common small-effect susceptibility variants in the vicinity of SCN5A and HEY2 providing evidence in support of this complex inheritance.3
Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
CPVT is commonly inherited in an autosomal dominant manner. In 65% of CPVT probands, the disorder is caused by a mutation in RYR2, resulting in CPVT type 1. A rarer form, CPVT2, is caused by recessive mutations in CASQ2 and is estimated to be present in 3–5% of patients with CPVT.1
Other genes involved in calcium homeostasis, including CALM1 and TRDN56, have also been implicated as a cause of CPVT. Mutations in CALM2 have been described in patients with overlapping features of LQTS and CPVT.9 Mutations in ANK2 and KCNJ2 that also cause, respectively, Ankyrin B syndrome (initially called LQT4) and Andersen–Tawil syndrome have been described in a few patients with normal QTc and exercise-induced arrhythmias.11,18 Furthermore, recessive mutations in TECRL have been associated with a clinical phenotype that has overlapping features of LQTS and CPVT.4
Long QT Syndrome (LQTS)
LQTS is most commonly inherited as an autosomal dominant disorder (initially known as Romano–Ward syndrome). To date, a number of genes have been implicated in LQTS. Genetic testing for LQTS has a diagnostic yield of ~60–70%.6
Mutations in KCNQ1, KCNH2 and SCN5A cause LQTS type 1 (LQT1), LQT2 and LQT3, respectively. Together, these subtypes account for ~75% of the patients with LQTS who have been genotyped (30–35% with mutations in KCNQ1, 25–40% in KCNH2 and ~5–10% in SCN5A).20 About 5% to 10% of patients with LQTS have multiple mutations in these genes, and symptoms typically present at a younger age and with a more severe phenotype in these patients than in patients with a single mutation. 21
Approximately 15–20% of patients with a definite clinical diagnosis of LQTS remain genotype-negative after extensive genetic testing.1 Single-nucleotide variants, small insertions or small deletions are the most commonly encountered disease-causing variations in these genes.19 However, large gene rearrangements have also been described.7
Short QT Syndrome (SQTS)
SQTS is inherited as an autosomal dominant disorder. Mutations in three genes KCNH2, KCNQ1 and KCNJ2, have been implicated in the disorder and are associated with its subtypes SQT1, SQT2 and SQT3, respectively.16 In contrast to loss-of-function variants associated with LQTS, SQTS-causing variants lead to a gain-of-function defect of the affected potassium channel. The genetic testing diagnostic yield in SQTS is low (<15%) and each known gene account for <5% of the patients with SQTS.10
Genetic testing of hereditary arrhythmias
Recommendations
The 2011 Heart Rhythm Society/European Heart Rhythm Association1 recommends:
- Genetic testing in patients with a clinical index of suspicion of one of the disorders
- Cascade genetic testing for family members and appropriate relatives of genotype-positive patients with any of these conditions
- Genetic testing of patients with a strong clinical index of suspicion for long QT syndrome, idiopathic QT prolongation with no symptoms and with documented electrocardiographic QTc interval >480 milliseconds (prepuberty) or >500 milliseconds (adults), or a clinical index of suspicion for catecholaminergic polymorphic ventricular tachycardia
- Genetic testing is not recommended for patients with isolated type 2 or type 3 Brugada electrocardiographic patterns, which are not diagnostic for Brugada syndrome
- For cardiac arrest survivors: testing of patients with a clinical index of suspicion for specific cardiomyopathy or channelopathy; otherwise, testing not indicated
- For postmortem testing in sudden unexpected death cases: the collection of tissue samples for possible future testing is recommended, and the selection of specific testing panels should be guided by autopsy findings (when available), and findings from clinical testing of surviving family members
Diagnostic Algorithm
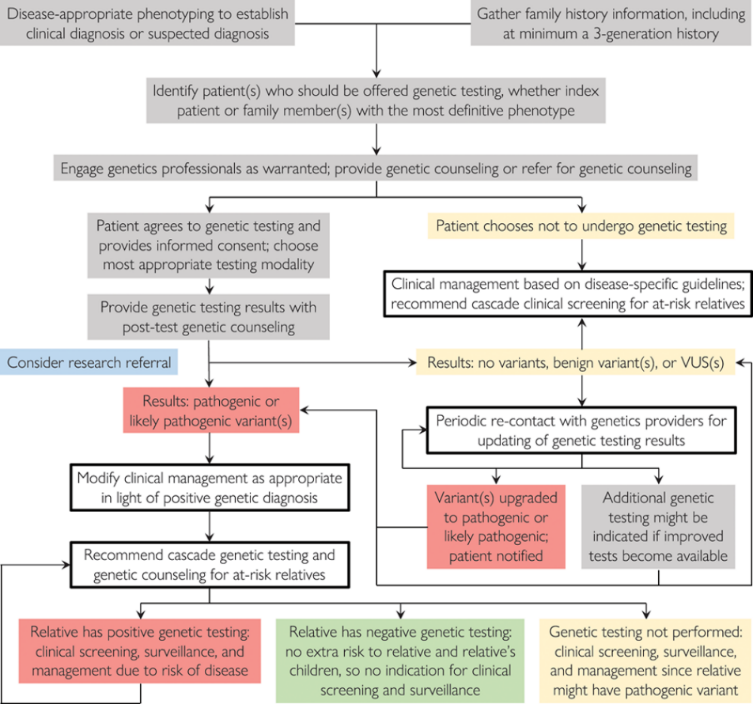
Musunuru et al.12 outlined strategies for genetic testing for patients with a confirmed or suspected diagnosis of a hereditary cardiac disorder. The result is the following diagnostic scheme:
Diagnosis and management
The diagnosis is based on clinical presentation and history, the characteristics of the electrocardiographic recording at rest and during exercise and genetic analyses.16 Management relies on pharmacological therapy, mostly β-adrenergic receptor blockers (specifically, propranolol and nadolol) and sodium and transient outward current blockers (such as quinidine), or surgical interventions, including left cardiac sympathetic denervation and implantation of a cardioverter–defibrillator.
Case studies
Brugada syndrome (BrS) case study
THE STORY, CASE PRESENTATION13
A 20-months-old boy was admitted to a hospital emergency service after his collapse during cycling. His father had a history of syncope at the age of 30. The fathers ajmaline challenge test was negative, but an ECG taken during a fever episode showed a BrS coved-shape ECG pattern. The sister of the father was diagnosed with an ajmaline-induced BrS type-1 ECG pattern.
THE TEST, ACTIONS TAKEN
ECG was performed and based on the results, he was diagnosed with sick sinus syndrome (SSS) with junctional escape. A DDD-pacemaker was implanted, and during the implantation, the dilated atria were electrically inactive, leading to the diagnosis of atrial standstill.
During recovery, he experienced a collapse anew, caused by a thrombus which led to a left-sided brain stroke. The echocardiography showed left atrial spontaneous echocardiographic contrast but no obvious thrombi. Although during follow-up no ventricular tachyarrhythmias were documented, an ECG taken during fever (39°C) unmasked the characteristic BrS type-1 pattern, and the boy received an endovascular VVI-implantable cardioverter-defibrillator as there is a high estimated risk of developing ventricular arrhythmias.
Genetic testing identified two SCN5A variants: c.4813+3_4813+6dupGGGT, a Belgian founder mutation, and c.4711T>C p. (Phe1571Leu). A familial segregation analysis revealed the founder mutation in the proband’s father and paternal aunt and de novo occurrence of the p.Phe1571Leu variant in the index patient.
THE OUTCOME, POST GENETIC TEST ACTIONS
BrS is rarely observed in the pediatric population, and only 4.3% of symptomatic patients experience their first malignant ventricular arrhythmic event before the age of 16 years. When SCN5A is affected by two compound variants, this can result in a significant aggravation of the disease severity and/or earlier disease onset. This was also observed in this patient who experienced his first syncope during physical activity at around the age of 2 years, probably caused by brady-arrhythmia in the setting of atrial standstill.
Clinical interventions mostly encompass the implantation of a pacemaker, with additional low-dose aspirin or oral quinidine, and single patient received β-blockers without pacemaker implantation.
In the presented family, heterozygous carriers of the SCN5A Belgian founder mutation showed characteristic BrS type-1 ECG pattern either after sodium channel blocker challenge (aunt) or during fever conditions (father) but presented no symptoms (aunt) or syncope at the age of 30 years (father).
Based on functional experiments, the described p.Phe1571Leu variant is likely pathogenic and, in the presented case, its de novo occurrence, together with the SCN5A Belgian founder mutation, explains the severe phenotype of cardiac sodium channelopathy with BrS.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) case study
THE STORY, CASE PRESENTATION17
A 17-year-old Caucasian boy was admitted to the intensive care unit (ICU) after successful resuscitation by emergency services. While performing running exercise in a fitness center, he suddenly collapsed. He had no symptoms prior to the collapse. However, in the years before, he had syncopated several times while climbing stairs, playing soccer, and once when he got frightened. A general practitioner previously performed an exercise ECG, which showed multiple premature beats under submaximal stress. As a result, beta-blockers were prescribed (metoprolol succinate 47.5 mg once per day). Apart from this, the patient had no medical history or prior medication. The patient was a nonsmoker with no regular alcohol consumption and an unremarkable family, social, and environmental history.
THE TEST, ACTIONS TAKEN
The diagnostic workup included blood tests, coronary angiography, electrophysiological testing, and cardiac magnetic resonance imaging, but all results were normal. Because the patient’s medical history included recurrent syncope during physical and emotional stress, CPVT was strongly suspected.
Genetic testing was performed and confirmed the diagnosis of CPVT, revealing a new heterozygous point mutation in the gene for RyR2, c.12520T>A (p.F4174 l, exon 90).
THE OUTCOME, POST GENETIC TEST ACTIONS
CPVT is an inherited disease presenting with recurrent syncope during exercise and acute emotions. This CPVT case is associated with a novel single point mutation in the RyR2 gene. 45% of the relevant mutations of RyR are located in this region.
Genetic screening of both parents showed that neither of them were carriers of the mutation. Results of further screening of the patient and family members for other pathogenic mutations (long or short QT syndrome, Brugada syndrome) were also negative.
The patient received an implantable cardioverter-defibrillator (ICD) device to protect him from recurrent episodes of ventricular fibrillation (VF). Additionally, oral medication with a beta-blocker was continued with the maximum tolerable dose. Shortly after implantation, the ICD terminated a sustaining ventricular tachycardia episode by anti-tachycardic pacing. This episode occurred early in the morning while the patient was asleep. In the following 6 months, a single episode of VF occurred during a physical activity (cycling), this time terminating spontaneously.
Long QT syndrome (LQTS) case study
THE STORY, CASE PRESENTATION5
A 30-year-old woman was initially diagnosed with epilepsy but tilt testing showed a vasodepressive response. Midodrine was prescribed, but without clinical improvement. She was referred for cardiology consultation due to continuing recurrent syncope; she reported episodes of presyncope and syncope, occasionally preceded by palpitations and sometimes triggered by standing. There was no other relevant medical history; her family history included the sudden death of her mother at age 46.
THE TEST, ACTIONS TAKEN
No abnormalities detected on laboratory tests and transthoracic echocardiography revealed no significant alterations. The 12-lead electrocardiogram (ECG) performed at the cardiology consultation showed sinus rhythm and significant QTc prolongation (500–565 ms).
Genetic testing identified the c.785delG mutation in heterozygosity in exon 4 of the KCNH2 gene, which leads to the formation of a premature STOP codon in position 359 in the protein (p.Gly262AlafsX98). Two further mutations were found: c.535G>A, in heterozygosity, in exon 3 of the KCNQ1 gene (p.Gly179Ser), and c.3068G>A, in heterozygosity, in exon 17 of the SCN5A gene (p.Arg1023His).
THE OUTCOME, POST GENETIC TEST ACTIONS
LQTS is a rare hereditary disease in patients without structural heart disease that is associated with a high risk of malignant ventricular tachycardia (VT) and sudden death.
The patient presented mutations in three different genes. The p.Gly262AlafsX98 mutation in the KCNH2 gene has been described in patients with LQTS and is known to be a cause of the condition. The mutations p.Gly179Ser in KCNQ1 and p.Arg1023His in SCN5A have also been reported in LQTS.
In view of the result of the previous tilt test and the non-specific symptoms, it was decided to implant an event recorder. The patient reported several episodes of pre-syncope, which corresponded to periods of sinus tachycardia, and in one of these non-sustained polymorphic VT was recorded. It was accordingly decided to implant an implantable cardioverter-defibrillator. Around a month after the procedure remote monitoring recorded an appropriate shock for polymorphic VT. Her beta-blocker therapy was changed to propanolol.
Short QT syndrome (SQTS) case study
THE STORY, CASE PRESENTATION2
A 6-months-old boy was first screened after his father died of confirmed sudden cardiac death (SCD) due to ventricular fibrillation (VF). The 12-lead surface ECG revealed a shortened QTc < 320 ms at that time. The patient suffered from syncope at the age of 1 year.
The electrophysiological study was performed and an intravenous ICD was implanted at the age of 16 due to the strong family history of SCD. His latest adequate ICD discharge was at the age of 17 due to VF. At the age of 19, he had two inappropriate therapies due to T-wave oversensing. After adjustment of the sensing threshold and initiation of antiarrhythmic therapy with quinidine, there were no other episodes of inadequate shocks.
His father was 28 years old when he died of SCD. He never had an ECG, cardiac imaging, or genetic testing performed. It was reported that his paternal grandmother also had a shortened QTc. His father’s siblings all received cardiovascular examinations including TTE, and none of them had shortened QTc-intervals or other pathological findings.
THE TEST, ACTIONS TAKEN
Genetic testing identified a pathogenic missense mutation in KCNH2 c.1764C>G (p.Asn588Lys). This mutation has been predicted to be deleterious according to SIFT, probably damaging according to Polyphen 2, and it has been previously associated with SQTS-1. No other mutations in genes associated with SQTS or SCD were found.
THE OUTCOME, POST GENETIC TEST ACTIONS
The KCNH2 mutation of SQTS family 1 c.1764C>G has been described as a gain of function mutation of the hERG channel associated with SQTS-1.
The most recent episode of VF at age 17 was not exercise-related. In 2015, the intravenous ICD was replaced due to battery depletion. During follow-up, the patient had a lead fracture. A transvenous lead extraction was attempted, but was not successful due to severe adhesions in the superior vena cava. The lead was hence left in place and a subcutaneous ICD was implanted. The patient had normal TTE findings. His most recent ECG at age 28 revealed a QTc of 304 ms.
References
[1] Ackerman MJ et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011 Aug;8(8):1308-39. doi: 10.1016/j.hrthm.2011.05.020. PMID: 21787999. https://www.heartrhythmjournal.com/article/S1547-5271(11)00607-2/fulltext
[2] Akdis D et al. Multiple clinical profiles of families with the short QT syndrome. Europace. 2018 Jun 1;20(FI1):f113-f121. doi: 10.1093/europace/eux186. PMID: 29016797. https://academic.oup.com/europace/article/20/FI1/f113/3979526
[3] Bezzina CR et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013 Sep;45(9):1044-9. doi: 10.1038/ng.2712. Epub 2013 Jul 21. Erratum in: Nat Genet. 2013 Nov;45(11):1409. Borggrefe, Martin [added]; Schimpf, Rainer [added]. PMID: 23872634; PMCID: PMC3869788. https://www.nature.com/articles/ng.2712
[4] Devalla HD et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016 Dec 1;8(12):1390-1408. doi: 10.15252/emmm.201505719. PMID: 27861123; PMCID: PMC5167130. https://www.embopress.org/doi/full/10.15252/emmm.201505719
[5] Fernandes M et al. Long QT syndrome with mutations in three genes: A rare case. Rev Port Cardiol. 2015 May;34(5):359.e1-5. English, Portuguese. doi: 10.1016/j.repc.2014.10.004. Epub 2015 Apr 29. PMID: 25935074. https://www.revportcardiol.org/en-long-qt-syndrome-with-mutations-articulo-S217420491500077X
[6] Hofman N et al. Yield of molecular and clinical testing for arrhythmia syndromes: report of 15 years’ experience. Circulation. 2013 Oct 1;128(14):1513-21. doi: 10.1161/CIRCULATIONAHA.112.000091. Epub 2013 Aug 20. PMID: 23963746. https://www.ahajournals.org/doi/10.1161/circulationaha.112.000091
[7] Koopmann TT et al. Long QT syndrome caused by a large duplication in the KCNH2 (HERG) gene undetectable by current polymerase chain reaction-based exon-scanning methodologies. Heart Rhythm. 2006 Jan;3(1):52-5. doi: 10.1016/j.hrthm.2005.10.014. PMID: 16399053. https://www.heartrhythmjournal.com/article/S1547-5271(05)02174-0/fulltext
[8] Le Scouarnec S et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet. 2015 May 15;24(10):2757-63. doi: 10.1093/hmg/ddv036. Epub 2015 Feb 3. PMID: 25650408. https://academic.oup.com/hmg/article/24/10/2757/622868
[9] Makita N et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014 Aug;7(4):466-74. doi: 10.1161/CIRCGENETICS.113.000459. Epub 2014 Jun 10. PMID: 24917665; PMCID: PMC4140998. https://www.ahajournals.org/doi/10.1161/CIRCGENETICS.113.000459
[10] Mazzanti A et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014 Apr 8;63(13):1300-1308. doi: 10.1016/j.jacc.2013.09.078. Epub 2013 Nov 28. PMID: 24291113; PMCID: PMC3988978. https://www.sciencedirect.com/science/article/pii/S0735109713062827
[11] Mohler PJ et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004 Jun 15;101(24):9137-42. doi: 10.1073/pnas.0402546101. Epub 2004 Jun 3. PMID: 15178757; PMCID: PMC428486. https://www.pnas.org/content/101/24/9137.long
[12] Musunuru K et al. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2020 Aug;13(4):e000067. doi: 10.1161/HCG.0000000000000067. Epub 2020 Jul 23. PMID: 32698598. https://www.ahajournals.org/doi/full/10.1161/HCG.0000000000000067
[13] Nijak A et al. Compound Heterozygous SCN5A Mutations in Severe Sodium Channelopathy With Brugada Syndrome: A Case Report. Front Cardiovasc Med. 2020 Jul 24;7:117. doi: 10.3389/fcvm.2020.00117. PMID: 32850980; PMCID: PMC7396896. https://www.frontiersin.org/articles/10.3389/fcvm.2020.00117/full
[14] Offerhaus JA et al. Epidemiology of inherited arrhythmias. Nat Rev Cardiol. 2020 Apr;17(4):205-215. doi: 10.1038/s41569-019-0266-2. Epub 2019 Oct 3. PMID: 31582838. https://www.nature.com/articles/s41569-019-0266-2
[15] Probst V et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. 2009 Dec;2(6):552-7. doi: 10.1161/CIRCGENETICS.109.853374. Epub 2009 Sep 29. PMID: 20031634. https://www.ahajournals.org/doi/10.1161/CIRCGENETICS.109.853374
[16] Schwartz PJ et al. Inherited cardiac arrhythmias. Nat Rev Dis Primers. 2020 Jul 16;6(1):58. doi: 10.1038/s41572-020-0188-7. PMID: 32678103; PMCID: PMC7935690. https://www.nature.com/articles/s41572-020-0188-7
[17] Seidlmayer LK et al. Description of a novel RyR2 mutation in a juvenile patient with symptomatic catecholaminergic polymorphic ventricular tachycardia in sleep and during exercise: a case report. J Med Case Rep. 2018 Oct 9;12(1):298. doi: 10.1186/s13256-018-1825-6. PMID: 30296944; PMCID: PMC6176516. https://jmedicalcasereports.biomedcentral.com/articles/10.1186/s13256-018-1825-6
[18] Tester DJ et al. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm. 2006 Jul;3(7):800-5. doi: 10.1016/j.hrthm.2006.03.025. Epub 2006 Mar 28. PMID: 16818210. https://www.heartrhythmjournal.com/article/S1547-5271(06)01327-0/fulltext
[19] Tester DJ et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005 May;2(5):507-17. doi: 10.1016/j.hrthm.2005.01.020. PMID: 15840476. https://www.heartrhythmjournal.com/article/S1547-5271(05)00191-8/fulltext
[20] Tester DJ et al. Effect of clinical phenotype on yield of long QT syndrome genetic testing. J Am Coll Cardiol. 2006 Feb 21;47(4):764-8. doi: 10.1016/j.jacc.2005.09.056. Epub 2006 Jan 26. PMID: 16487842. https://www.sciencedirect.com/science/article/pii/S0735109705028007
[21] Westenskow P et al. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004 Apr 20;109(15):1834-41. doi: 10.1161/01.CIR.0000125524.34234.13. Epub 2004 Mar 29. PMID: 15051636. https://www.ahajournals.org/doi/10.1161/01.CIR.0000125524.34234.13