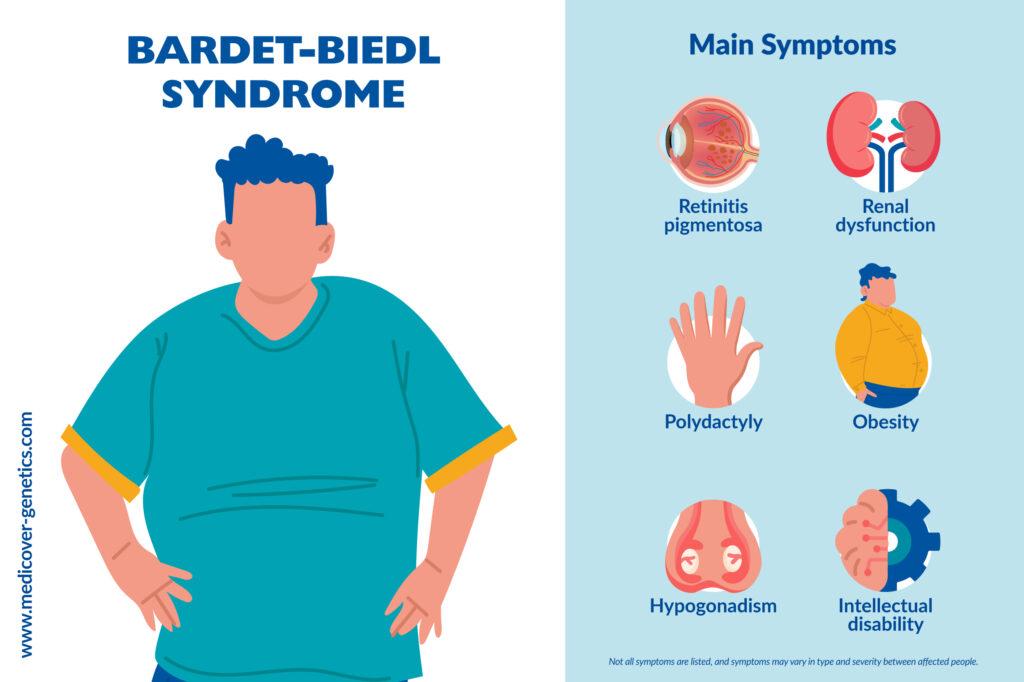
Bardet-Biedl syndrome is a genetic disorder that affects multiple organs of the body, especially the kidney. It is a ciliopathy in which retinitis pigmentosa, renal dysfunction and polydactyly are observed in combination with obesity, hypogonadism, and behavioral abnormalities. It is caused due to genetic changes (mutations) in more than 20 different genes.
Also called
Bardet-Bielt Syndrome is also known as:
- BBS
Symptoms
Symptoms vary from person to person and may include:
- Retinitis pigmentosa
- Renal dysfunction
- Polydactyly
- Obesity
- Hypogonadism
- Behavioral abnormalities
Frequency
Bardet-Biedl syndrome occurs with a prevalence of about 1:25,000.
Causes
Bardet-Biedl syndrome is usually caused by autosomal recessive genetic mutations, and more than 20 BBS genes and modifier genes have been identified.
More than two mutations in more than one gene locus may be causative, so that in addition to recessive inheritance of two variants in one gene, a further variant in another BBS gene can act as a modifier and contribute to the clinical expression of the disease.
In about 60-80% of clinically diagnosed cases, genetic changes in the previously known BBS genes are detected, with the genes BBS1 and BBS10 being most frequently affected in Europeans (23% and 20% of cases, respectively).
Inheritance
The genetic causes of Bardet-Biedl syndrome are usually autosomal recessive, although triallelic inheritance has also been described.
Differential diagnosis
Syndromes with similar symptoms to Bardet-Biedl syndrome include Laurence-Moon syndrome (LMS), Alström syndrome, Meckel (Meckel-Gruber) syndrome, McKusick-Kaufman syndrome (MKKS), Biemon II syndrome, and Prader-Willi syndrome
Alström syndrome is a rare disease that shows phenotypic similarities to BBS and is caused by autosomal recessive genetic changes in the ALMS1 gene. The symptoms of Alström syndrome include retinitis pigmentosa, obesity, kidney and liver dysfunction, insulin resistance and hyperinsulinemia, and dilated cardiomyopathy.
Treatment
Treatment for Bardet-Biedl syndrome may include:
- Surgeries to correct physical abnormalities
- Kidney transplantation
- Active lifestyle to combat obesity
- Use of setmelanotide, an approved treatment for chronic weight management for BBS patients
References
Mary, Laura et al. “Bardet-Biedl syndrome: Antenatal presentation of forty-five fetuses with biallelic pathogenic variants in known Bardet-Biedl syndrome genes.” Clinical genetics vol. 95,3 (2019): 384-397. doi:10.1111/cge.13500, https://onlinelibrary.wiley.com/doi/10.1111/cge.13500.
Niederlova, Veronika et al. “Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes.” Human mutation vol. 40,11 (2019): 2068-2087. doi:10.1002/humu.23862, https://onlinelibrary.wiley.com/doi/10.1002/humu.23862.
Manara, Elena et al. “Mutation profile of BBS genes in patients with Bardet-Biedl syndrome: an Italian study.” Italian journal of pediatrics vol. 45,1 72. 13 Jun. 2019, doi:10.1186/s13052-019-0659-1, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6567512/.
Suspitsin, Evgeny N, and Evgeny N Imyanitov. “Bardet-Biedl Syndrome.” Molecular syndromology vol. 7,2 (2016): 62-71. doi:10.1159/000445491, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4906432/.
Khan, S A et al. “Genetics of human Bardet-Biedl syndrome, an updates.” Clinical genetics vol. 90,1 (2016): 3-15. doi:10.1111/cge.12737, https://onlinelibrary.wiley.com/doi/10.1111/cge.12737.
Priya, Sathya et al. “Bardet-Biedl syndrome: Genetics, molecular pathophysiology, and disease management.” Indian journal of ophthalmology vol. 64,9 (2016): 620-627. doi:10.4103/0301-4738.194328, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5151149/.
Novas, Rossina et al. “Bardet-Biedl syndrome: Is it only cilia dysfunction?.” FEBS letters vol. 589,22 (2015): 3479-91. doi:10.1016/j.febslet.2015.07.031, https://febs.onlinelibrary.wiley.com/doi/10.1016/j.febslet.2015.07.031.
Schmidts, Miriam. “Clinical genetics and pathobiology of ciliary chondrodysplasias.” Journal of pediatric genetics vol. 3,2 (2014): 46-94. doi:10.3233/PGE-14089, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4262788/.
Barker, Amy R et al. “Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development.” Organogenesis vol. 10,1 (2014): 96-107. doi:10.4161/org.27375, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4049900/.
Romani, Marta et al. “Joubert syndrome: congenital cerebellar ataxia with the molar tooth.” The Lancet. Neurology vol. 12,9 (2013): 894-905. doi:10.1016/S1474-4422(13)70136-4, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3809058/.
Ronquillo, C C et al. “Senior-Løken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis.” Vision research vol. 75 (2012): 88-97. doi:10.1016/j.visres.2012.07.003, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3504181/.
Brugmann, Samantha A et al. “Craniofacial ciliopathies: A new classification for craniofacial disorders.” American journal of medical genetics. Part A vol. 152A,12 (2010): 2995-3006. doi:10.1002/ajmg.a.33727, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3121325/.
“Bardet-Biedl Syndrome - Symptoms, Causes, Treatment | NORD.” Rarediseases.org, www.rarediseases.org/rare-diseases/bardet-biedl-syndrome. Accessed 13 Dec. 2023.
“Bardet-Biedl Syndrome: MedlinePlus Genetics.” Medlineplus.gov, ww.medlineplus.gov/genetics/condition/bardet-biedl-syndrome/. Accessed 13 Dec. 2023.