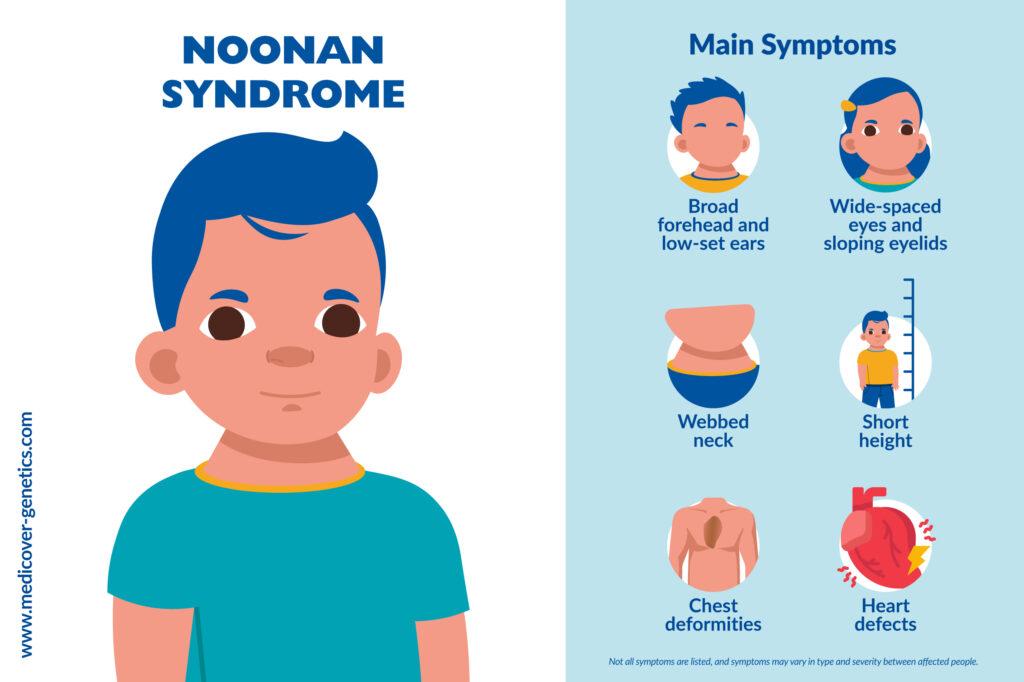
Noonan syndrome is an autosomal dominant inherited disorder with a broad, variable spectrum of symptoms, including facial dysmorphia, short stature, chest deformities and heart defects. It belongs to a group of cardiovascular conditions known as RASopathies, and in 50% of cases is due to pathogenic variants in the PTPN11 gene.
Also called
Noonan Syndrome is also known as:
- NS
- Familial Turner syndrome
- Female pseudo-Turner syndrome
- Male Turner syndrome
- Noonan’s syndrome
- Noonan-Ehmke syndrome
- Pseudo-Ullrich-Turner syndrome
- Turner phenotype with normal karyotype
- Turner syndrome in female with X chromosome
- Turner-like syndrome
- Ullrich-Noonan syndrome
Symptoms
Noonan syndrome has a lot of symptoms that can differ in range and severity from person to person and can depend on the mutation that causes the syndrome.
Symptoms may include:
- Facial dysmorphia (broad forehead, wide-spaced eyes, low-set ears, sloping eyelids)
- Proportional short stature
- Pterygium colli (webbed neck)
- Chest deformities
- Cryptorchidism (absence of at least one testicle)
- Mental retardation
- A slight tendency to bleeding disorders
- Heart defects, usually in the form of pulmonary stenosis or hypertrophic cardiomyopathy
Frequency
The frequency of Noonan syndrome is estimated to be 1 in 1000 to 2500 live births. Noonan syndrome is more common in males than females.
Causes
The PTPN11 gene, which codes for a cytoplasmic protein tyrosine phosphatase (SHP-2), is the gene mainly affected in Noonan syndrome. Pathogenic variants in the PTPN11 gene, which almost exclusively lead to amino acid exchanges, are the molecular cause in about 50% of all Noonan patients investigated so far.
Pathogenic variants have currently been identified in several other genes, including the RAS‑ERK-MAP kinase signal transduction in Noonan syndrome. Mutations in the SOS1 (Son of Sevenless) gene were detected in up to 15% of Noonan patients who did not have a mutation in the PTPN11 gene. Pathogenic variants in the RAF1 gene could be identified in about 8% of Noonan patients. Almost all patients with RAF1 mutations show hypertrophic cardiomyopathy. Other genes include RIT1 (approximately 5% of patients) and KRAS (approximately 3% of patients), pathogenic variants here predominantly lead to severe phenotypes.
Pathogenic variants in the genes MAP2K1, MAP2K2 and BRAF are identified less frequently in Noonan syndrome, although these genes are more frequently altered in patients with cardiofaciocutaneous (CFC) syndrome. The incidence of pathogenic BRAF variants in Noonan syndrome is estimated to be 2%. Here, the patients described show neonatal growth retardation, slight cognitive deficits and muscle hypotonia. Even rarer are variants in the NRAS and CBL genes (1%). In addition, other genes described in connection with Noonan syndrome include RASA2, PPP1CB (Noonan syndrome-like disorder with loose anagen hair), and MRAS.
Variants of these genes and genes MAP2K2, SHOC2, HRAS and NF1 are also found in patients with Noonan syndrome-like diseases, such as CFC syndrome, LEOPARD syndrome, Noonan syndrome-like disorder with juvenile myelomonocytic leukemia (JMML), and neurofibromatosis-Noonan syndrome, among others. Pathogenic variants in the NF1 gene have also been detected in Noonan syndrome patients who do not fulfill the clinical criteria for classic neurofibromatosis (NF).
In about 25% of all patients with Noonan syndrome, no causative variant can be detected in the genes that are currently known.
Inheritance
Noonan syndrome is inherited in an autosomal dominant pattern, meaning that if one parent carries a mutated copy of a gene that causes Noonan syndrome, it can lead to Noonan syndrome in the offspring.
Differential diagnosis
Syndromes with similar symptoms to Noonan syndrome include Cardiofaciocutaneous (CFC) syndrome, Turner syndrome, Costello syndrome, Neurofibromatosis-Noonan syndrome, and Noonan syndrome with multiple lentigines (formerly known as LEOPARD syndrome).
Treatment
Multidisciplinary treatment is required, from cardiologists, hematologists and endocrinologists, among other healthcare professionals. Early interventions may help and be beneficial for individuals with Noonan syndrome.
Treatment may include:
- Surgical interventions for heart defects
- Medications for heart defects
- Medications or supportive measures for individuals with thrombocytopenia and to prevent respiratory infections
- Remedial education
- Speech therapy
- Physical therapy
References
Motta, Marialetizia et al. “Activating MRAS mutations cause Noonan syndrome associated with hypertrophic cardiomyopathy.” Human molecular genetics vol. 29,11 (2020): 1772-1783. doi:10.1093/hmg/ddz108, https://academic.oup.com/hmg/article/29/11/1772/5492387?login=false.
El Bouchikhi, Ihssane et al. “Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate.” International journal of pediatrics & adolescent medicine vol. 3,4 (2016): 133-142. doi:10.1016/j.ijpam.2016.06.003, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6372459/.
Chen, Peng-Chieh et al. “Next-generation sequencing identifies rare variants associated with Noonan syndrome.” Proceedings of the National Academy of Sciences of the United States of America vol. 111,31 (2014): 11473-8. doi:10.1073/pnas.1324128111, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4128129/.
Vissers, Lisenka E L M et al. “Heterozygous germline mutations in A2ML1 are associated with a disorder clinically related to Noonan syndrome.” European journal of human genetics: EJHG vol. 23,3 (2015): 317-24. doi:10.1038/ejhg.2014.115, Epub 2014 Jun 18, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4326711/.
Aoki, Yoko et al. “Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome.” American journal of human genetics vol. 93,1 (2013): 173-80. doi:10.1016/j.ajhg.2013.05.021, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3710767/.
Tartaglia, Marco et al. “Noonan syndrome and clinically related disorders.” Best practice & research. Clinical endocrinology & metabolism vol. 25,1 (2011): 161-79. doi:10.1016/j.beem.2010.09.002, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3058199/.
Tartaglia, M et al. “Noonan syndrome: clinical aspects and molecular pathogenesis.” Molecular syndromology vol. 1,1 (2010): 2-26. doi:10.1159/000276766, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2858523/pdf/msy0001-0002.pdf.
Cirstea, Ion C et al. “A restricted spectrum of NRAS mutations causes Noonan syndrome.” Nature genetics vol. 42,1 (2010): 27-9. doi:10.1038/ng.497, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3118669/.
Sarkozy, Anna et al. “Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum.” Human mutation vol. 30,4 (2009): 695-702. doi:10.1002/humu.20955, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4028130/.
Nava, Caroline et al. “Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome.” Journal of medical genetics vol. 44,12 (2007): 763-71. doi:10.1136/jmg.2007.050450, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2652823/.
Razzaque, M Abdur et al. “Germline gain-of-function mutations in RAF1 cause Noonan syndrome.” Nature genetics vol. 39,8 (2007): 1013-7. doi:10.1038/ng2078, https://www.nature.com/articles/ng2078.
Roberts, Amy E et al. “Germline gain-of-function mutations in SOS1 cause Noonan syndrome.” Nature genetics vol. 39,1 (2007): 70-4. doi:10.1038/ng1926, https://www.nature.com/articles/ng1926.
Carta, Claudio et al. “Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype.” American journal of human genetics vol. 79,1 (2006): 129-35. doi:10.1086/504394, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1474118/.
Schubbert, Suzanne et al. “Germline KRAS mutations cause Noonan syndrome.” Nature genetics vol. 38,3 (2006): 331-6. doi:10.1038/ng1748, https://www.nature.com/articles/ng1748.
Noonan, J A. “Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease.” American journal of diseases of children (1960) vol. 116,4 (1968): 373-80. doi:10.1001/archpedi.1968.02100020377005.
“Noonan Syndrome: MedlinePlus Genetics.” Medlineplus.gov, 1 June 2018, www.medlineplus.gov/genetics/condition/noonan-syndrome
Noonan Syndrome - Symptoms, Causes, Treatment | NORD.” Rarediseases.org, www.rarediseases.org/rare-diseases/noonan-syndrome/