Wagner syndrome is a very rare autosomal dominantly inherited degenerative eye disease with complete penetrance. Symptoms, such as a reduction in visual acuity and cataract formation that often leads to blindness, begin around the age of 20. The genetic cause of Wagner syndrome is pathogenic variants in the VCAN gene.
Also called
Wagner Syndrome is also known as:
- Hyaloideoretinal degeneration of Wagner
- VCAN-related vitreoretinopathy
- Wagner disease
- Wagner vitreoretinal degeneration
- Wagner vitreoretinopathy
Symptoms
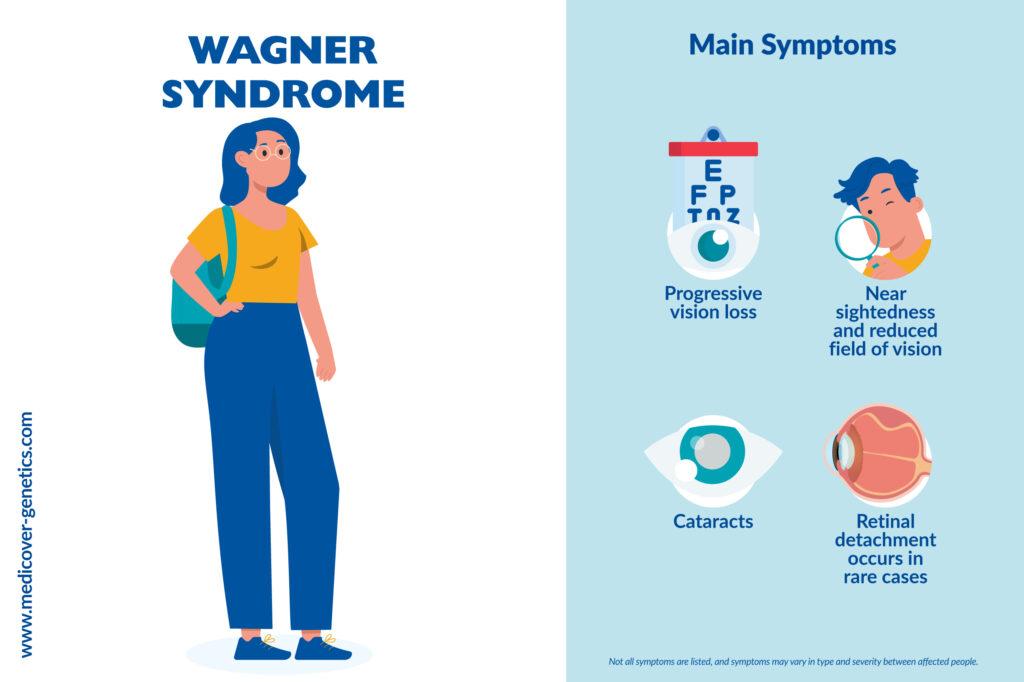
Symptoms begin around the age of 20 and are characterized by:
- Progressive vision loss
- Visual field restriction
- Moderate myopia
- Cataract formation that usually leads to blindness
- Morphological characteristics such as optically empty vitreous cavity, fibrillar condensations and chorioretinal dystrophy
- Retinal detachment occurs in rare cases
Frequency
The frequency of Wagner syndrome is unknown due to its rarity. Around 300 people with Wagner syndrome have been described, with approximately half of them originating from the Netherlands.
Causes
The genetic cause of Wagner syndrome appears to be heterogeneous. In 26 families examined by molecular genetic methods so far, the cause was found to be in variants in the VCAN gene in seven families. VCAN codes for the chondroitin sulfate proteoglucan, Versican, a structural protein of the vitreous body. So far, all variants have been localized in the splice regions of exon 8.
Inheritance
Wagner syndrome is inherited in an autosomal dominant pattern, meaning that if one parent carries a mutated copy of the VCAN gene, it can lead to Wagner syndrome in the offspring.
Differential diagnosis
In some cases, it is difficult to differentiate Wagner syndrome from Stickler syndrome. In Wagner syndrome, however, facial and skeletal malformations are absent.
Other syndromes with similar symptoms to Wagner syndrome include Goldmann-Favre disease, familial exudative vitreoretinopathy, and autosomal dominant vitreoretinochoroidopathy (ADVIRC).
Treatment
Treatment for Wagner syndrome may include:
- Ophthalmologic examinations
- Prophylactic laser or cryocoagulation
- Scleral buckling surgery
- Vitrectomy
- Phacoemulsification and implantation of an artificial intraocular lens to treat cataracts
References
Ankala, Arunkanth et al. “Is exon 8 the most critical or the only dispensable exon of the VCAN gene? Insights into VCAN variants and clinical spectrum of Wagner syndrome.” American journal of medical genetics. Part A vol. 176,8 (2018): 1778-1783. doi:10.1002/ajmg.a.38855, https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.38855.
Kloeckener-Gruissem, Barbara et al. “Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of Wagner syndrome.” European journal of human genetics: EJHG vol. 21,3 (2013): 352-6. doi:10.1038/ejhg.2012.137, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3573191/pdf/ejhg2012137a.pdf.
Ronan, Shawn M et al. “Mutational hot spot potential of a novel base pair mutation of the CSPG2 gene in a family with Wagner syndrome.” Archives of ophthalmology (Chicago, Ill. : 1960) vol. 127,11 (2009): 1511-9. doi:10.1001/archophthalmol.2009.273, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3514888/.
Meredith, Sarah P et al. “Clinical characterisation and molecular analysis of Wagner syndrome.” The British journal of ophthalmology vol. 91,5 (2007): 655-9. doi:10.1136/bjo.2006.104406, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1954774/.
Mukhopadhyay, Arijit et al. “Erosive vitreoretinopathy and wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants.” Investigative ophthalmology & visual science vol. 47,8 (2006): 3565-72. doi:10.1167/iovs.06-0141, https://iovs.arvojournals.org/article.aspx?articleid=2126518.
Kloeckener-Gruissem, Barbara et al. “Identification of the genetic defect in the original Wagner syndrome family.” Molecular vision vol. 12 350-5. 17 Apr. 2006, http://www.molvis.org/molvis/v12/a39/.
Wagner Syndrome: MedlinePlus Genetics.” Medlineplus.gov, www.medlineplus.gov/genetics/condition/wagner-syndrome/. Accessed 14 Dec. 2023.
“Orphanet: Wagner Disease.” Orphanet, https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=89/. Accessed 14 Dec. 2023.