Smith-Magenis syndrome (SMS) is a global developmental delay disorder that affects several organs of the body. It is caused by a microdeletion on the short arm of chromosome 17 (17p11.2). The main characteristics of SMS are distinct facial features, sleep disturbances, behavioral problems, and delayed speech and language development.
Also called
Smith-Magenis Syndrome is also known as:
- Chromosome 17, interstitial deletion 17p
- Chromosome 17p11.2 deletion syndrome
- SMCR
- Smith-Magenis chromosome region
- Retinoic acid induced 1 gene (RAI1)
- SMS
Symptoms
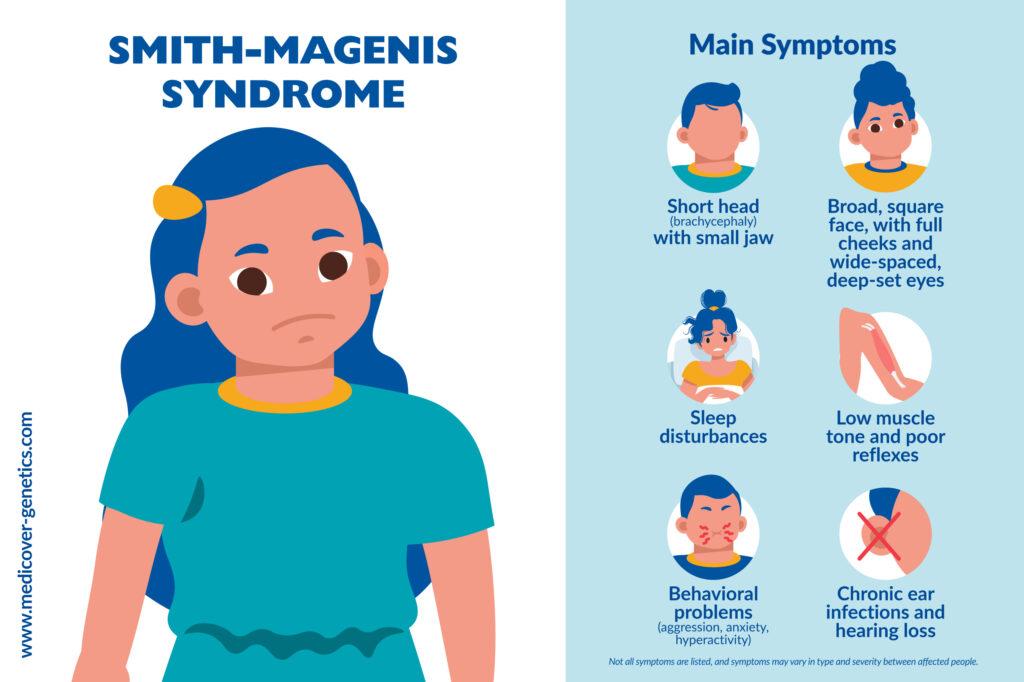
SMS has a lot of symptoms that can differ from person to person. Discrete facial features and typical behaviors are the main features of SMS. Facial characteristics can often be diagnosed in the early stages of life.
Distinctive facial features include:
- Broad, square-shaped face with deep-set eyes
- Full cheeks
- Brachycephaly (short head)
- Small jaw (micrognathia)
- Prominent lower jaw develops with age
- Flat midface
- Broad nasal bridge
- M-shaped curved upper lip with distinct philtrum
- Down-turned upper lip
- Hypertelorism (wide-set eyes)
Other symptoms include:
- Low, hoarse voice
- Forearm limitations in infancy and early childhood
- Hypotonia
- Hyporeflexia
- Feeding difficulties
- Sleep disturbances
- Behavioral problems such as aggression, anxiety, hyperactivity, tantrums and self-injury
- Hearing loss in up to 70% of patients as a result of chronic middle ear infections
- Myopia (nearsightedness) in approximately in 50% of patients, occasionally a detached retina
- Lordosis (curved lumbar spine) and scoliosis (sideways curvature of the spine)
- Foot deformities
- Congenital heart and kidney malformations in about 30% of patients
Many children with SMS are described as musical and most are good with computers, which can be used for support.
Frequency
The frequency of SMS is estimated to be 1 in 15,000 to 25,000 individuals worldwide. Both men and women are equally impacted by SMS. However, many cases may go undiagnosed or misdiagnosed.
Causes
SMS is caused by a microdeletion of variable size (ranging from 1.5 to 9 Mb, usually about 3.7 Mb) on the short arm of chromosome 17 (17p11.2), which includes the RAI1 gene, in 90% of cases and by a pathogenic variant in the RAI1 gene in 10% of cases. The RAI1 gene is responsible for most of the symptoms of SMS. An additional 13 genes located in the deleted region modify the severity (contiguous gene syndrome).
Inheritance
Between 70 and 80% cases of SMS are caused by a de novo (new) deletion that occurs spontaneously during embryonic development and can occur in individuals with no family history of SMS. Consequently, the risk of recurrence is low if a parental chromosomal structural change in chromosome 17 or a rare parental mosaic for a RAI1 variant has been ruled out. The remaining 10-15% are due to the inheritance of a variant in the RAI1 gene from an unaffected parent who is a carrier of the disorder.
Differential diagnosis
Large deletions can be found with conventional chromosomal analysis, and the more common small ones with FISH analysis or a chromosomal microarray.
Syndromes with similar symptoms to SMS include Down syndrome, Williams syndrome (Williams-Beuren syndrome), Prader-Willi syndrome, Angelman syndrome, Sotos syndrome, Fragile X syndrome, DiGeorge syndrome (chromosome 22q11.2 deletion syndrome), 9q34 deletion syndrome (Kleefstra syndrome), 2q37 deletion syndrome, 2q23.1 deletion syndrome, 1p36 deletion syndrome.
Treatment
Treatment for SMS is tailored to the individual’s specific symptoms and may include:
- Speech/language therapy
- Physical therapy
- Occupational therapy
- Behavioral therapy
- Medications for sleep disturbances
- Educational support
References
Ann CM Smith, et al. “Smith-Magenis Syndrome.” Nih.gov, University of Washington, Seattle, 20 June 2019, www.ncbi.nlm.nih.gov/books/NBK1310/.
Nag, Heidi Elisabeth et al. “Parental experiences with behavioural problems in Smith-Magenis syndrome: The need for syndrome-specific competence.” Journal of intellectual disabilities vol. 23,3 (2019): 359-372, doi:10.1177/1744629519847375, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6734585/.
Goldenberg, Paula. “An Update on Common Chromosome Microdeletion and Microduplication Syndromes.” Pediatric annals vol. 47,5 (2018): e198-e203, doi:10.3928/19382359-20180419-01, https://journals.healio.com/doi/full/10.3928/19382359-20180419-01.
Elsea, Sarah H, and Santhosh Girirajan. “Smith-Magenis syndrome.” European journal of human genetics vol. 16,4 (2008): 412-21. doi:10.1038/sj.ejhg.5202009, https://www.nature.com/articles/5202009.
Rost, I. “Chromosomale Mikro- Deletionssyndrome.” Monatsschrift Kinderheilkunde, vol. 148, no. 1, 26 Jan. 2000, pp. 55–69, https://doi.org/10.1007/s001120050015.
Greenberg, F et al. “Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2).” American journal of medical genetics vol. 62,3 (1996): 247-54. doi:10.1002/(SICI)1096-8628(19960329)62:3<247::AID-AJMG9>3.0.CO;2-Q.
“Smith-Magenis Syndrome: MedlinePlus Genetics.” Medlineplus.gov, www.medlineplus.gov/genetics/condition/smith-magenis-syndrome. Accessed 07 Dec. 2023.
“Smith Magenis Syndrome - Symptoms, Causes, Treatment | NORD.” Rarediseases.org, www.rarediseases.org/rare-diseases/smith-magenis-syndrome/#disease-overview-main.